






Current methods for the studies of protein 3-D structures include X-ray crystallography, nuclear magnetic resonance (NMR), and electron microscopy. Protein X-ray crystallography requires a balanced integration of the cumulative results of many experiments.
The processes involved in protein structure analysis are summarized below.
cDNA Cloning and Construction of Expression Plasmids

Protein production

The proteins required for analysis are produced using an expression plasmid, either in living cells such as E. coli or in reconstituted cell-free systems. A large amount of protein is produced using optimum conditions, determined through extensive testing, which allow for the efficient production of correctly folded proteins. Genetic engineering and protein engineering techniques are essential for achieving the expression and easy purification of the target proteins.
Protein Purification

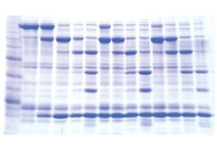
Target proteins are purified from a mixture of many different proteins based on differences in physical and chemical properties and structure using column chromatography under appropriate conditions (confirmation of the target protein purification can be made by methods such as SDS-PAGE).
Protein Crystallization


In crystallization, proteins are arrayed in a regular 3-D array. Proteins grow as crystals under certain conditions. A large number of conditions must be met in order to yield high-quality crystals. Thus, development of automated crystallization robot systems is an important challenge for allowing for more efficient experiments.
Diffraction images from crystals and diffraction data collection



When high-quality protein crystals, with the proteins in a regular array, are irradiated with X-rays, the electrons surrounding the atoms that make up the proteins scatter the X-rays. The X-rays interfere with one another, producing diffraction images. High quality crystals and images obtained through intense low-emission X-rays are required for high-precision analysis. The SPring-8 beamlines provide stable, high-quality X-rays suitable for the crystal structure analysis of proteins.
Identification of protein structures
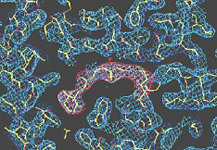
Based on the collected diffraction data, the phases of the reflections are determined using methods such as multi-wavelength anomalous diffraction (MAD), enabling calculation of the electron distribution (electron density map). The electron density shows the shape of the molecule. A protein structure model is then created using 3-D computer graphics, and a computer performs further calculations to refine the model in greater detail. In the figure, the electron density is shown as a mesh of light blue and red lines, and the precisely-created protein model is shown inside the yellow mesh.
Accumulation of 3-D Structure Information
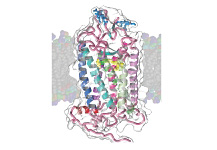
Expansion of the database will allow rough structure estimations based on proteins with similar amino acid sequences. In the future, the accumulated 3-D structure information and further development and improvement in model creation software, along with the continuing evolution of computers, will enable the creation of models for existing individual proteins in high precision, without the need for actual experiments.

Research center of synchrotron radiation science that uses SPring-8.
1-1-1, Kouto, Sayo-cho, Sayo-gun, Hyogo JAPAN 679-5148 RIKEN SPring-8 Center.
Copyright © RIKEN SPring-8 Center.